Description
PeptideAtlas collects raw mass
spectrometry proteomics datasets from laboratories around the world and reprocesses them in a
uniform bioinformatics workflow using the
Trans-Proteomic Pipeline .
This track displays peptide identifications from the PeptideAtlas
August 2014 (Build 433) Human build.
This build, based on 971 samples containing 420,607,360 spectra, identified 1,021,823 distinct
peptides, covering 15,136 canonical proteins.
Each PeptideAtlas build comprises a set of reprocessed experiments from a single species or subset of samples (such has human plasma) from a species. Processed results are filtered to a quality level such that there is a 1% false discovery rate at the protein level. All peptide identifications of sufficient quality to enter a build are mapped to the Ensembl genome (v75) using the Ensembl toolkit. Genomic coordinates for all identified peptides to all their Ensembl protein, transcript, and gene mappings, including intron spans, as calculated by the Ensembl toolkit are stored in the PeptideAtlas database.
All peptide sequences in the August 2014 human build (including unmapped sequences) are available for
download in FASTA format.
Methods
Mass spectrometer spectra are compared to theoretical spectra (SEQUEST, X!Tandem) or actual
spectra (SpectraST) to identify possible peptides.
These peptide identifications are scored and filtered (using PeptideProphet) to retain
only the highest scoring identifications.
The filtered sequences are compared to protein sequence
databases (for human, Ensembl, IPI, and Swiss-Prot).
The CDS coordinates relative to protein start of matched sequences are used to
then calculate genomic coordinates.
The protein identifications are then clustered and annotated using ProteinProphet,
and stored in the SBEAMS database, where they
assigned a unique identifer of the form PAp[8 digit number], e.g. PAp00000001.
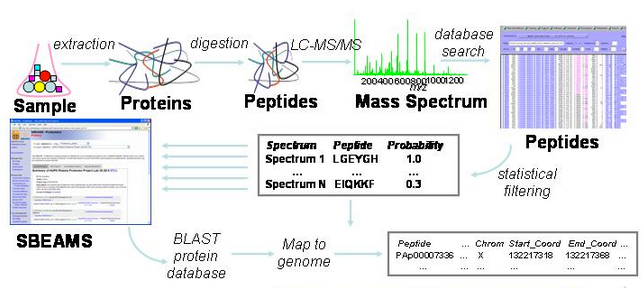
The processing pipeline is summarized in the graphic below.
Credits
Eric Deutsch, Zhi Sun, and the PeptideAtlas team at the Institute for Systems Biology, Seattle.
References
Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN,
Aebersold R.
The PeptideAtlas project.
Nucleic Acids Res. 2006 Jan 1;34(Database issue):D655-8.
PMID: 16381952; PMC: PMC1347403
Farrah T, Deutsch EW, Omenn GS, Sun Z, Watts JD, Yamamoto T, Shteynberg D, Harris MM, Moritz RL.
State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and
plasma proteomes for the biology- and disease-driven Human Proteome Project.
J Proteome Res. 2014 Jan 3;13(1):60-75.
PMID: 24261998; PMC: PMC3951210
Keller A, Nesvizhskii AI, Kolker E, Aebersold R.
Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and
database search.
Anal Chem. 2002 Oct 15;74(20):5383-92.
PMID: 12403597
Nesvizhskii AI, Keller A, Kolker E, Aebersold R.
A statistical model for identifying proteins by tandem mass spectrometry.
Anal Chem. 2003 Sep 1;75(17):4646-58.
PMID: 14632076
|
|