Description
This track contains the location and score of transcription factor
binding sites conserved in the human/mouse/rat alignment. A binding
site is considered to be conserved across the alignment if its score
meets the threshold score for its binding matrix in all 3 species.
The score and threshold are computed with the Transfac Matrix Database (v7.0) created by
Biobase.
The data are purely computational, and as such not all binding sites
listed here are biologically functional binding sites.
In the graphical display, each box represents one conserved putative tfbs. Clicking on
a box brings up detailed information on the binding site, namely its
Transfac I.D., a link to its Transfac Matrix (free registration with Transfac
required), its location in the human genome (chromosome, start, end,
and strand), its length in bases, its raw score, and its Z score.
All binding factors that are known to bind to the particular binding matrix
of the binding site
are listed along with their species, SwissProt ID, and a link to that
factor's page on the UCSC Protein Browser if such an entry exists.
Methods
The Transfac Matrix Database (v.7.0) contains position-weight matrices for
398 transcription factor binding sites, as characterized through
experimental results in the scientific literature. Only binding matrices
for known transcription factors in human, mouse, or rat were used for this
track (258 of the 398). A typical (in this
case ficticious) matrix (call it mat) will look something like:
A C G T
01 15 15 15 15 N
02 20 10 15 15 N
03 0 0 60 0 G
04 60 0 0 0 A
05 0 0 0 60 T
The above matrix specifies the results of 60 (the sum of each row)
experiments. In the experiments, the first position of the binding site
was A 15 times, C 15 times, G 15 times, and T 15 times (and so on for
each position.) The consensus sequence of the above binding site as
characterized by the matrix is NNGAT. The format of the consensus sequence
is the deduced consensus in the IUPAC 15-letter code.
In the general case, the goal is to find all matches to a matrix of length n
that are conserved across ns sequences. For this example, n=5 and
ns=3 (human, mouse, and rat.) Denote the multispecies alignment s,
such that sji is the nucleotide at position j of species i. Also,
define an ns x 4 background matrix (call it back) giving the background
frequencies of each nucleotide in each species. A sliding window (of length n)
calculates the "species score" for each species at each position:
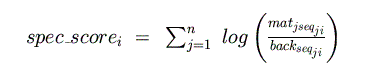
From this, a log-odds score is calculated for each species (normalizing by the
length of the matrix and the number of species in the alignment):
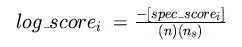
These scores are then summed for all species, yielding a final log-odds score for
the current position:
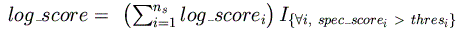
Note that the log-odds score of each species must exceed the threshold for that
species. The threshold is calculated for each species such that the only hits
that will be reported will have a Z score (to be discussed later) of 1.64 or
higher in each species (corresponding to a p-value of 0.05). Next, the maximum
and minimum possible log-odds scores
are computed and summed across all species for the given binding matrix:
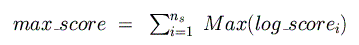
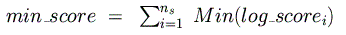
These are then used to normalize the final, raw log-odds score so that its range is
between 0 and 1:
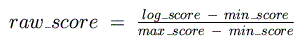
Next, the best raw score for each binding matrix is calculated for the 5,000 base
upstream region of each human RefSeq gene (taken from the RefGene table for hg19.)
The mean and standard deviation for each binding matrix are then calculated across
all RefSeq genes. These are then used to create the threshold for each binding matrix,
namely, 1.64 standard deviations above the mean. Tfloc is then run with this threshold
on each chromosome for the 3-way multiz alignments. Finally, a Z score is calculated
for each binding site hit h to matrix m according to the following formula:
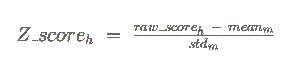
This final Z score can be interpreted as the number of standard deviations above the
mean raw score for that binding matrix across the upstream regions of all RefSeq genes.
The default Z score cutoff for display in the browser is 2.33 (corresponding to a p-value
of 0.01.) This cutoff can be adjusted at the top of this page.
After all hits have been recorded genome-wide, one final filtering step is performed.
Due to the inherant redundancy of the Transfac database, several binding sites that
all bind the same factor often appear together. For example, consider the following
binding sites:
585 chr1 4021 4042 V$MEF2_02 875 - 2.83
585 chr1 4021 4042 V$MEF2_03 917 - 3.38
585 chr1 4021 4042 V$MEF2_04 844 - 3.45
585 chr1 4022 4037 V$HMEF2_Q6 810 - 2.34
585 chr1 4022 4037 V$MEF2_01 802 - 2.47
585 chr1 4022 4038 V$RSRFC4_Q2 875 - 2.65
585 chr1 4022 4039 V$AMEF2_Q6 823 - 2.44
585 chr1 4023 4038 V$RSRFC4_01 878 + 2.53
585 chr1 4024 4035 V$MEF2_Q6_01 913 + 2.41
585 chr1 4024 4039 V$MMEF2_Q6 861 - 2.39
These 10 overlapping binding sites bind a total of 19 factors. However,
of these 19 factors, only 7 of them are unique. Many of the above
binding sites are redundant (they add no additional factors). In fact, the first
3 binding sites all bind the same two factors (namely, aMEF-2 and MEF-2A). These ten binding
sites can therefore be filtered down to the following four binding sites, without any
loss of information (in terms of transcription factors). The final table entry
then has the following four lines, since these four binding sites account for
all 7 of the unique factors:
585 chr1 4021 4042 V$MEF2_04 844 - 3.45
585 chr1 4022 4038 V$RSRFC4_Q2 875 - 2.65
585 chr1 4024 4035 V$MEF2_Q6_01 913 + 2.41
585 chr1 4024 4039 V$MMEF2_Q6 861 - 2.39
In the event that multiple binding sites bind the same factors, the site with
the highest Z score is chosen. Only binding sites which overlap each other
and whose start positions are within 5 bases of each other are considered for
merging.
It should be noted that the positions of many of these conserved binding
sites coincide with known exons and other highly conserved regions.
Regions such as these are more likely to contain false positive matches,
as the high sequence identity across the alignment increases the likelihood of
a short motif that looks like a binding site to be conserved. Conversely,
matches found in introns and intergenic regions are more likely to be real
binding sites, since these regions are mostly poorly conserved.
These data were obtained by running the program tfloc (Transcription Factor binding
site LOCater) on multiz46way alignments, restricting only to the July 2007 (mm9) mouse genome assembly, the November 2004 rat assembly (rn4), and the February 2009 human genome assembly (hg19).
Transcription factor information was culled from the Transfac Factor
database, version 7.0.
Table Format
The format of the tfbsConsSites sql table is shown above.
The columns are (from left to right): bin, chromosome, from, to, binding matrix ID, raw score,
strand, and Z score.
To get the corresponding transcription factor information for a given binding matrix, use the table
tfbsConsFactors. The format of the tfbsConsFactors sql table is:
V$MYOD_01 M00001 mouse MyoD P10085
V$E47_01 M00002 human E47 N
V$CMYB_01 M00004 mouse c-Myb P06876
V$AP4_01 M00005 human AP-4 Q01664
V$MEF2_01 M00006 mouse aMEF-2 Q60929
V$MEF2_01 M00006 rat MEF-2 N
V$MEF2_01 M00006 human MEF-2A Q02078
V$ELK1_01 M00007 human Elk-1 P19419
V$SP1_01 M00008 human Sp1 P08047
V$EVI1_06 M00011 mouse Evi-1 P14404
The columns are (from left to right): transfac binding matrix id,
transfac binding matrix accession number, transcription factor species,
transcription factor name, SwissProt accesssion number.
When no factor species, name, or id information exists in the transfac factor
database for a binding matrix, an 'N' appears in the corresponding column(s). Notice also
that if more than one transcription factor is known for one binding matrix, each occurs on its own line,
so multiple lines can exist for one binding matrix.
Credits
These data were generated using the Transfac Matrix and Factor databases created by
Biobase.
The tfloc program was developed at The Pennsylvania State University (with numerous
updates done at UCSC) by Matt Weirauch.
This track was created by Matt Weirauch and Brian Raney at The
University of California at Santa Cruz.
|
|